How “Biased” GPCR Drugs Could Revolutionize Medicine—With Fewer Side Effects
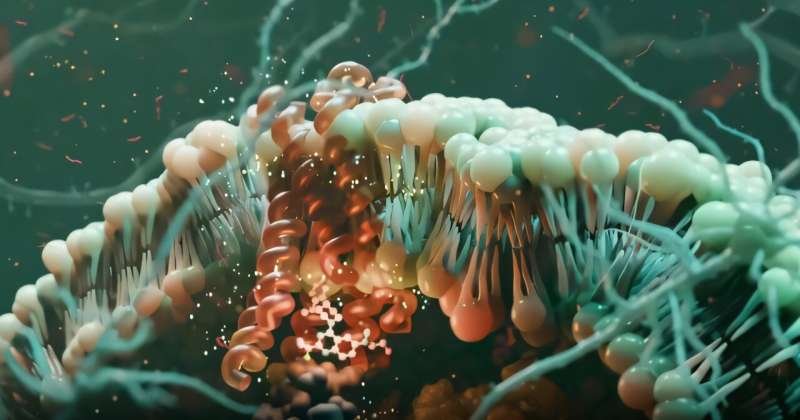
Beyond One-Size-Fits-All: The Rise of Precision GPCR Drugs
For decades, drugs targeting G protein-coupled receptors (GPCRs) have been medical mainstays—powering treatments for everything from high blood pressure and allergies to depression and pain. In fact, about one-third of all FDA-approved drugs act on this massive family of cell surface receptors.
But traditional GPCR drugs often come with a catch: unwanted side effects. That’s because they activate all downstream pathways at once—like flipping a master switch that controls both healing and harm.
Now, a breakthrough from Sanford Burnham Prebys, the University of Minnesota, and Duke University is changing the game. Published in Nature (October 2025), their research reveals how “biased” signaling molecules can selectively activate only the therapeutic arms of GPCR pathways—while blocking the ones that cause side effects.
Even better: they’ve discovered a predictable, rational way to design these smarter drugs by targeting the inside of the cell.
What Are GPCRs—and Why Do They Matter?
GPCRs are molecular antennas embedded in cell membranes. They detect signals outside the cell—like hormones, neurotransmitters, or light—and relay that information inside by activating two key protein families:
- G proteins (which trigger rapid cellular responses)
- Beta-arrestins (which regulate receptor activity and drive other signaling cascades)
With 826 known GPCRs in humans—and roles in nearly every physiological process—they’re prime drug targets. Yet only ~165 are currently “drugged.” Why?
Many GPCRs lack clear binding pockets on their outside surface, making them seem “undruggable.”
“But the intracellular side always binds G proteins or beta-arrestin,” says Dr. Steven Olson of Sanford Burnham Prebys. “So there’s almost always a pocket there.”
The Problem with Traditional GPCR Drugs
Most existing GPCR drugs bind the extracellular side of the receptor and activate all downstream pathways equally—what scientists call “balanced agonism.”
This can be problematic. For example:
- A painkiller might relieve discomfort but also suppress breathing (via one G protein)
- A psychiatric drug might improve mood but cause dangerous drops in blood pressure (via another)
The solution? Biased agonists—compounds that favor only the beneficial pathways.
“If we can control signaling bias,” explains Dr. Lauren Slosky (University of Minnesota), “we can separate efficacy from toxicity.”
A New Strategy: Targeting the Inside of the Cell
The research team focused on the neurotensin receptor 1 (NTSR1), a GPCR linked to addiction, schizophrenia, and pain—but historically hard to drug due to severe side effects like hypothermia and low blood pressure.
They studied a compound called SBI-553, a biased modulator that binds inside the cell—not outside.
Using structural biology and functional assays, they discovered something remarkable:
SBI-553 doesn’t just turn pathways on or off—it fine-tunes them.
Specifically, it acts as:
- A “molecular bumper” that blocks certain G proteins (like Gq, linked to side effects)
- A “molecular glue” that promotes binding of other G proteins or beta-arrestins that drive therapeutic effects
This isn’t all-or-nothing signaling—it’s a precise dial, not a switch.
Small Changes, Big Impact
The team tested 29 analogs of SBI-553—each with tiny chemical tweaks. The results were striking:
- Minor structural changes dramatically altered which G proteins were activated or inhibited
- One analog, SBI-593, failed to block Gq—and failed to prevent hypothermia in mice
- SBI-553, which fully inhibits Gq, avoided this side effect entirely
“Small differences in the molecule lead to big differences in biology,” says Dr. Slosky.
Critically, these effects were predictable—a first in biased GPCR drug design.
Why This Changes Drug Discovery
For years, biased GPCR drugs were seen as unpredictable and hard to engineer. But this study proves that intracellular allosteric modulators offer a rational path forward.
Benefits of this approach: ✅ Targets “undruggable” GPCRs with no external binding pockets
✅ Reduces side effects by silencing harmful pathways
✅ Enables precision medicine for neurological, metabolic, and immune disorders
✅ Accelerates development through structure-based design
“We’ve shown this intracellular site is druggable,” says Dr. Olson. “And the interactions are predictable—making it viable for real-world drug discovery.”
Relevance to Clinical Microbiology & Therapeutics
While GPCRs are often associated with neuroscience or cardiology, they also play key roles in immunology and infection:
- Immune cells use GPCRs to detect chemokines during inflammation
- Some pathogens hijack host GPCRs to enter cells or evade immunity
- GPCR-targeting drugs are used to treat HIV (maraviroc), allergies (antihistamines), and sepsis-related shock
Understanding biased signaling helps clinicians:
- Choose drugs with cleaner safety profiles
- Anticipate off-target effects
- Support the development of next-generation anti-inflammatories or immunomodulators
Final Thought: From Blunt Tools to Surgical Precision
The era of “one receptor, one drug, all pathways” is ending. With intracellular biased modulators, we’re entering an age of pathway-selective pharmacology—where drugs don’t just hit a target, but orchestrate a precise cellular response.
As Dr. Slosky puts it:
“We’re no longer stuck with the receptor’s default settings. We can reprogram it.”
For patients, that could mean more effective treatments with fewer risks—a win for medicine, microbiology, and human health alike.
Stay Ahead in Molecular Therapeutics
At ClinicalMicrobiology.org, we bridge basic science and clinical practice—covering breakthroughs in drug design, host-pathogen interactions, and cellular signaling.
👉 Subscribe today for expert insights on emerging therapies, resistance mechanisms, and the future of precision medicine.
Note: This article is based on original research published in Nature (October 2025) by Sanford Burnham Prebys, University of Minnesota, and Duke University. Scientific credit belongs to Dr. Lauren Slosky, Dr. Steven Olson, and colleagues. Content has been fully rewritten for clarity, originality, and educational value.





Post Comment